Ingest + Export¶
--workflow ingest_export
Downloads full Seurat objects from LabKey, a URL, or a local filepath for each sample and immediately exports the raw RNA count matrix as a 10x-like directory. No GPU, no HPC required — runs on a local Mac or any Linux host.
Stage-by-stage dataflow¶
flowchart TD
SS["**samplesheet.csv**
sample_id · output_file_id · url · path · species"]
DISPATCH{"**Tri-mode dispatch**
Which column is non-empty?"}
INGEST_LABKEY["**INGEST_LABKEY**
(Rdiscvr)
Downloads from LabKey via output_file_id"]
INGEST_URL["**INGEST_URL**
(Rdiscvr)
Downloads from public URL"]
INGEST_FILE["**INGEST_FILE**
(Rdiscvr)
Loads from local filepath"]
EXPORT_COUNTS["**EXPORT_COUNTS**
(CellMembrane)
Extracts raw counts → 10x-like matrix dir"]
OUT_RDS["outputs/ingest/{sample_id}.rds
Full Seurat object"]
OUT_COUNTS["outputs/counts/{sample_id}_counts/
matrix.mtx · features.tsv
barcodes.tsv · obs_meta.csv"]
SS --> DISPATCH
DISPATCH -->|"output_file_id"| INGEST_LABKEY
DISPATCH -->|"url"| INGEST_URL
DISPATCH -->|"path"| INGEST_FILE
INGEST_LABKEY -->|"tuple(meta, .rds)"| OUT_RDS
INGEST_URL -->|"tuple(meta, .rds)"| OUT_RDS
INGEST_FILE -->|"tuple(meta, .rds)"| OUT_RDS
INGEST_LABKEY -->|"tuple(meta, .rds)"| EXPORT_COUNTS
INGEST_URL -->|"tuple(meta, .rds)"| EXPORT_COUNTS
INGEST_FILE -->|"tuple(meta, .rds)"| EXPORT_COUNTS
EXPORT_COUNTS -->|"tuple(meta, counts_dir/)"| OUT_COUNTSInputs¶
Samplesheet¶
Path: --input (default data/samplesheet.csv)
Each row must have exactly one of output_file_id, url, or path populated. See Data Formats → Samplesheet for the full column specification.
sample_id,output_file_id,url,path,species
SAMPLE_LABKEY,12345,,,human
SAMPLE_URL,,https://example.org/data.rds,,macaque
SAMPLE_FILE,,,/home/user/data/mydata.h5ad,mouse
Required parameters (LabKey mode)¶
| Parameter | Description |
|---|---|
--labkey_base_url |
LabKey server base URL |
--labkey_folder |
LabKey folder path |
These parameters are only required for rows that use output_file_id (LabKey mode). Rows using url or path do not need LabKey credentials.
Optional parameters¶
| Parameter | Default | Description |
|---|---|---|
--export_assay |
RNA |
Seurat assay to export as count matrix |
--outdir |
outputs/ |
Output directory |
Outputs¶
INGEST → outputs/ingest/{sample_id}.rds¶
A full Seurat RDS object (counts + all metadata), produced identically by all three ingest modules. Contains at minimum the cells passing QC and their RNA assay.
| File | Description |
|---|---|
{sample_id}.rds |
Full Seurat object downloaded from LabKey, URL, or loaded from local path |
EXPORT_COUNTS → outputs/counts/{sample_id}_counts/¶
A 10x-like matrix directory compatible with e.g. Seurat::Read10X(), scanpy.read_10x_mtx(), or BPCells::open_matrix_dir():
| File | Description |
|---|---|
matrix.mtx |
Sparse raw count matrix in Market Exchange (MatrixMarket) format. Rows = genes, columns = cells. |
features.tsv |
Gene names, one per row, matching row order in matrix.mtx. |
barcodes.tsv |
Cell barcodes, one per row, matching column order in matrix.mtx. |
obs_meta.csv |
Cell-level metadata from seurat_object[[]] with additional columns sample_id, species, output_file_id, and barcode. |
Synthetic example export¶
The docs and CI use a seeded fixture bundle in tests/fixtures/synthetic_trial_data/ so the exported count layout is visible without any live Prime-seq download.
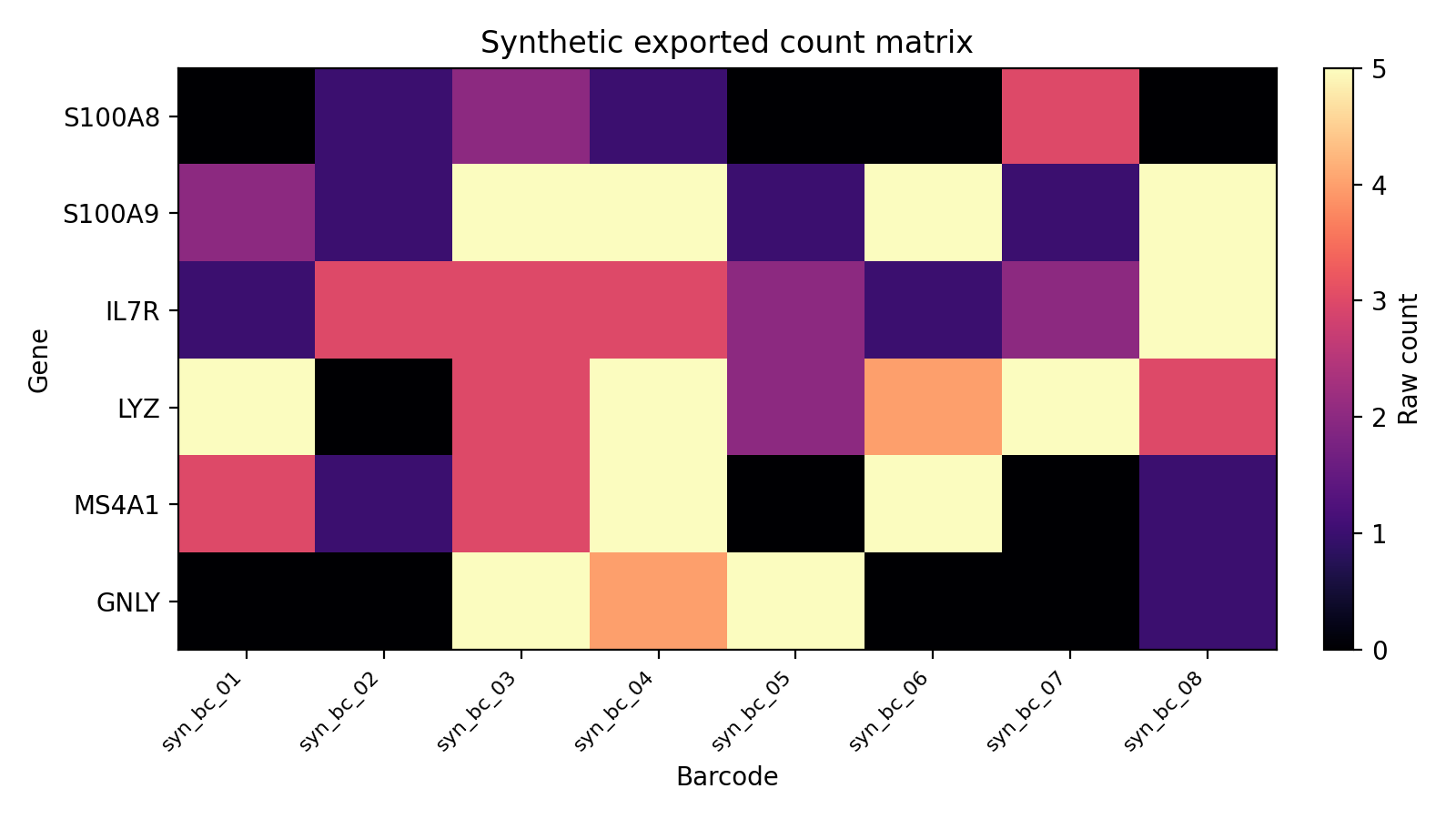
This is the same file shape produced by EXPORT_COUNTS: matrix.mtx, features.tsv, barcodes.tsv, and obs_meta.csv.
For the generated code-level reference, see API Reference → Workflows.
Running locally¶
LabKey mode¶
nextflow run main.nf \
--workflow ingest_export \
--labkey_base_url https://labkey.example.org \
--labkey_folder /My/Project/Folder
URL mode (no LabKey required)¶
Local file mode (no LabKey required)¶
On macOS (or Linux without SLURM) the local executor is auto-selected; no -profile flag is required.
To limit output location:
nextflow run main.nf \
--workflow ingest_export \
--outdir ./outputs/dev \
--labkey_base_url https://labkey.example.org \
--labkey_folder /My/Project/Folder
Running on HPC¶
For routine SLURM runs, the recommended entrypoint is a copied runs/<name>/run.sh template. The command below shows the repo-root launcher alternative.
bash slurm_nextflow.sh \
--workflow ingest_export \
--labkey_base_url https://labkey.example.org \
--labkey_folder /My/Project/Folder
Container prerequisites: When running on SLURM, Apptainer must be configured before your first run. See Container image pre-pull and SIF cache in the usage guide for graphroot setup, storage details, and all
NXF_APPTAINER_*environment variables.
Resource profile¶
| Step | CPUs | Memory | Wall time |
|---|---|---|---|
| INGEST_LABKEY / INGEST_URL / INGEST_FILE | 4 | 32 GB | 4 h |
| EXPORT_COUNTS | 4 | 32 GB | 4 h |